Written by:
Catherine Overed-Sayer
Associate Director, Bioscience COPD/IPF at AstraZeneca
Uncovering genetic insights that hold a key to transforming disease remain a crucial part of the drug discovery process being undertaken by our scientists across the globe. One particular area of high unmet need we are working to address is in idiopathic pulmonary fibrosis (IPF), a chronic, progressive lung disease which leads to scarring (fibrosis) of the lungs, respiratory failure and death. Currently only two treatments are approved to slow down disease progression in IPF. More needs to be done to improve patient outcomes and extend survival rates beyond their current figure of three to five years from diagnosis.1
Although there is evidence of a genetic predisposition to IPF in some patients, knowledge of the specific genes that are responsible for disease development and progression remains limited.2 Through our pioneering R&D we are seeking to improve understanding of the complex disease biology of IPF, identifying underlying drivers of disease and finding new genetic targets for developing therapies that we hope could modify and one day maybe even cure this debilitating disease.
Uncovering how IPF develops
One of the main challenges when discovering novel treatments for IPF is that the biological mechanisms driving disease progression are still somewhat unclear. Over recent years the scientific community has learned more about the biology of IPF. We now know that the epithelial cells of the alveoli that form part of the gas exchange barrier between the lung parenchyma and the blood undergo recurrent injury and an aberrant wound healing response. This ‘out of control’ healing response activates cells known as fibroblasts to deposit an extracellular matrix comprised of collagen, resulting in fibrosis of the lung tissue. On high resolution CT scans this matrix resembles a ‘honeycomb’ pattern within the lungs, impairing gas exchange and making it harder for patients to breathe.
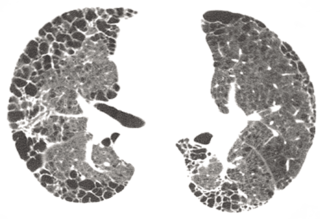
Image Source: Arch Pathol Lab Med.2012;136(6):591–600. Image courtesy of Richard Webb, MD.
Some patients with IPF progress rapidly and others more slowly, however predicting how patients will progress is very challenging, and the identified genetic differences between these patients are actually quite limited. Additionally, some patients appear to be more susceptible to experiencing disease exacerbations which can be extremely serious as patients often rapidly lose a substantial proportion of their lung function – sadly even one or two exacerbations can be fatal.3
The current therapies used to treat IPF slow the disease, but don’t halt its progression, and certainly don’t reverse the lung scarring or cure the disease. We are trying to target several different cellular mechanisms that we believe may be pivotal in IPF progression including:
- Inhibiting fibroblast activation
- Modulating the detrimental effect of cell ageing (senescence) on fibrosis
- Regenerative biology, where we’re trying to restore epithelial cell function in the lungs.
One thing that we are sure of is that the biology of IPF is broad, affecting multiple pathways and a range of different cell types. We have an array of potential targets in our pipeline and although we hope our late-stage assets will be successful, we are also trying to identify further novel disease pathways to target.
We have an investigational treatment in Phase 1 clinical trials which may provide a new approach to target fibrotic pathways earlier versus other mechanisms in development for IPF. Our porcupine inhibitor has been shown to suppress Wnt signalling (an important pathway in cell maintenance, differentiation and renewal), which is known to be highly active in diseases like IPF. By suppressing targeted signalling pathways, we hope that the novel treatment may be able to slow or prevent fibrosis in IPF and potentially other types of fibrotic diseases.
Seeking novel gene targets
Getting down to the genetic level of IPF is essential if we want to target the underlying cause of the disease. If we can learn more about the genetic interplays in IPF, we will be better placed to identify patient subtypes who are more likely to progress rapidly, as well as those that may or may not respond well to treatment. Our hope is that these genetic associations will provide clues into the mechanisms that may be causally associated with IPF, some of which the scientific community may already be exploring, and some that may not have been identified before. This type of Precision Medicine approach is now being applied across 90% of our R&D pipeline.
No one has really nailed down exactly which genes are the most instrumental in IPF, but our knowledge continues to grow as more genome-wide genetic association studies confirm new disease targets. Recently, findings were published from the largest IPF whole exome sequencing study which confirmed that a novel gene known as KIF15 is associated with IPF.4,5 The results, which were published in the American Journal of Respiratory and Critical Care Medicine, showed that variations in the KIF15 gene lead to decreased protein expression and reduced cell proliferation, increasing susceptibility to IPF.5
This type of genetic understanding is helping us to better comprehend the fundamentals of IPF, transforming drug discovery as we strive to find new ways to treat, prevent, modify, and eventually even cure the disease.
Advancing disease understanding in collaboration
I firmly believe that scientific discovery is more effective when done in collaboration. We partner with a broad range of different groups to progress IPF disease understanding and advance treatment. We actively seek expertise and insights through both academic and industry partnerships and ensure that we always keep patients front-of-mind. We achieve this by working closely with physicians based in hospitals and laboratories, as well as IPF patients who are the experts on living with the disease – their biology and genetics will help us to progress scientific understanding and discover future targeted medicines ultimately for their benefit.
A particularly exciting collaboration is between AstraZeneca and BenevolentAI where we’re using artificial intelligence to create ‘living’ maps of disease known as Knowledge Graphs.They enable us to analyse vast amounts of scientific data and literature to see potential interactions between gene targets, expression and disease. We can find previously unexplored patterns of disease and are able to draw better and faster conclusions across a range of complex diseases.
Watch this video to find out how Knowledge Graphs are helping researchers to identify interactions within chronic diseases:

The first time we ran Knowledge Graphs for IPF over 200 potential gene targets were revealed. Once triaged collaboratively with BenevolentAI, and using CRISPR screening and bespoke target validation such as cell-based in vitro assays, we reduced this down to fewer than 10 targets, helping us to focus our efforts on the gene targets that may have the greatest potential. Knowledge Graphs are accelerating how we do our science, making our drug discovery more efficient and helping us to get novel targeted treatments to patients quicker. As a result of our collaboration with BenevolentAI we have identified a novel gene target for IPF that has now been selected for our portfolio and will continue to be explored by our scientists across the globe.
In addition to understanding the genetic connections in IPF, we are also focusing on the biology of IPF as part of our early-stage R&D. Our PhD and postdoctoral researchers are working with academia to gain a better understanding of complex cellular mechanisms involving multiple cell types, and the role they play in IPF. They are utilising multicellular 3D in vitro assays such as precision cut lung slices and iPSC-derived alveolospheres, as well as exploring the role of extracellular vesicles (which deliver proteins, metabolites and nucleic acids to recipient cells) in IPF.
Aspiring to change the future of IPF
IPF is a challenging and multifactorial disease – there’s still so much that we don’t fully comprehend. I’m optimistic that over the next decade the research being undertaken by our scientists and others across academia and the pharmaceutical industry will improve our understanding of the genes involved in IPF and how they influence disease progression. There are a huge number of potential targets under investigation which provides hope that we will find effective treatments that can address the underlying cause of the disease, improving outcomes and changing the future for patients living with this devastating disease.